
כל תוכן iLive נבדק מבחינה רפואית או נבדק למעשה כדי להבטיח דיוק עובדתי רב ככל האפשר.
יש לנו קווים מנחים קפדניים המקור רק קישור לאתרים מדיה מכובד, מוסדות מחקר אקדמי, בכל עת אפשרי, עמיתים מבחינה רפואית מחקרים. שים לב שהמספרים בסוגריים ([1], [2] וכו ') הם קישורים הניתנים ללחיצה למחקרים אלה.
אם אתה סבור שתוכן כלשהו שלנו אינו מדויק, לא עדכני או מפוקפק אחרת, בחר אותו ולחץ על Ctrl + Enter.
מחלת שארקו-מארי-טות'.
המומחה הרפואי של המאמר

סקירה אחרונה: 12.07.2025
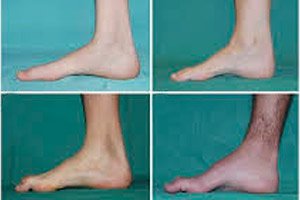
ניוון שרירים פרינאלי, תסמונת או מחלת שארקו-מארי-טות' היא קבוצה של מחלות תורשתיות כרוניות עם נזק לעצבים ההיקפיים.
לפי ICD-10, בסעיף הדן במחלות מערכת העצבים, הקוד למחלה זו הוא G60.0 (נוירופתיה מוטורית וחושית תורשתית). היא נכללת גם ברשימת המחלות היתומות.
אֶפִּידֶמִיוֹלוֹגִיָה
על פי נתונים סטטיסטיים קליניים, שכיחות כל סוגי מחלת שארקו-מארי-טות' לכל 100 אלף מהאוכלוסייה היא 19 מקרים (על פי מקורות אחרים, מקרה אחד לכל 2.5-10 אלף מהאוכלוסייה).
CMT סוג 1 מהווה כשני שלישים מהמקרים (מקרה אחד לכל 5,000 עד 7,000 תושבים), וכמעט 70% מהם קשורים לשכפול של הגן PMP22. יותר מ-1.2 מיליון איש ברחבי העולם סובלים מסוג זה של מחלה.
שכיחות CMT סוג 4 מוערכת ב-1-5 מקרים לכל 10,000 ילדים. [ 1 ]
גורם ל מחלת שארקו-מארי-טות'
על פי סיווג תסמונות פולינוירופתיות, ניוון שרירים פרינאלי (פיבולרי), אמיוטרופיה עצבית מסוג Charcot-Marie-Tooth או מחלת Charcot-Marie-Tooth (בקיצור CMT) מתייחסת לפולינוירופתיה מוטורית-חושית שנקבעה גנטית. [ 2 ]
כלומר, הגורמים להופעתה הם מוטציות גנטיות. ובהתאם לאופי הסטיות הגנטיות, נבדלים הסוגים או הסוגים העיקריים של תסמונת זו: דה-מיאלינציה ואקסונלית. הקבוצה הראשונה כוללת את מחלת Charcot-Marie-Tooth סוג 1 (CMT1), המתרחשת עקב שכפול של הגן PMP22 בכרומוזום 17, המקודד לחלבון מיאלין היקפי טרנסממברני 22. כתוצאה מכך, מתרחשת דה-מיאלינציה סגמנטלית של מעטפת האקסון (תהליכים של תאי עצב) וירידה במהירות הולכת אותות העצבים. בנוסף, מוטציות עשויות להיות בגנים אחרים.
הצורה האקסונלית היא מחלת Charcot-Marie-Tooth סוג 2 (CMT2), הפוגעת באקסונים עצמם וקשורה לשינויים פתולוגיים בגן MFN2 בלוקוס 1p36.22, המקודד לחלבון הממברנה מיטופוסין-2, הנחוץ לאיחוי המיטוכונדריה וליצירת רשתות מיטוכונדריה פונקציונליות בתוך תאי עצב היקפיים. ישנם יותר מתריסר תת-סוגים של CMT2 (עם מוטציות בגנים ספציפיים).
יש לציין כי כיום זוהו יותר ממאה גנים שנזקם, המועבר בתורשה, גורם לתת-סוגים שונים של מחלת שארקו-מארי-טות'. לדוגמה, מוטציות בגן RAB7 מובילות ל-CMT מסוג 2B; שינוי בגן SH3TC2 (המקודד לאחד מחלבוני הממברנה של תאי שוואן) גורם ל-CMT מסוג 4C, המתבטא בילדות ומאופיין בדה-מיאלינציה של נוירונים מוטוריים ותחושתיים (ישנן תריסר וחצי צורות של סוג 4 של מחלה זו).
תסמונת CMT מסוג 3 נדירה (הנקראת תסמונת דג'רין-סוטאס) מתחילה להתפתח בילדות המוקדמת ונגרמת על ידי מוטציות בגנים PMP22, MPZ, EGR2 וגנים אחרים.
כאשר מתרחשת CMT מסוג 5 בגיל 5-12 שנים, נצפית לא רק נוירופתיה מוטורית (בצורה של פרפרזיס ספסטי של הגפיים התחתונות), אלא גם נזק לעצבים האופטיים והשמיעתיים.
חולשת שרירים ואטרופיה של הראייה (עם אובדן ראייה) כמו גם בעיות שיווי משקל אופייניות ל-CMT מסוג 6. ובמחלת Charcot-Marie-Tooth מסוג 7 יש לא רק נוירופתיה מוטורית-חושית, אלא גם מחלת רשתית בצורת רטיניטיס פיגמנטוזה.
מחלת שרקו-מארי-טות' עם טטרפרזיס של הגפיים (חולשת תנועה של הזרועות והרגליים), הנפוצה יותר בקרב גברים, היא סוג של דה-מיאלינציה, ונחשבת כנובעת ממוטציה בגן GJB1 בזרוע הארוכה של כרומוזום X, המקודד לקונקסין 32, חלבון טרנסממברני של תאי שוואן ואוליגודנדרוציטים המווסת את העברת אותות עצביים. [ 3 ]
גורמי סיכון
גורם הסיכון העיקרי ל-CMT הוא היסטוריה משפחתית של המחלה, כלומר, אצל קרובי משפחה.
על פי גנטיקאים, אם שני ההורים נשאים של הגן האוטוזומלי הרצסיבי למחלת שארקו-מארי-טות', הסיכון לילד שיפתח מחלה זו הוא 25%. והסיכון שהילד יהיה נשא של גן זה (אך לא יהיו לו תסמינים כלשהם) מוערך ב-50%.
במקרה של תורשה מקושרת לכרומוזום X (כאשר הגן המוטנטי נמצא על כרומוזום X של האישה), קיים סיכון של 50% שהאם תעביר את הגן לבנה, אשר יפתח CMT. ייתכן שהמחלה לא תופיע כאשר ילדה תיוולד, אך בניה של הבת (נכדיה) עשויים לרשת את הגן הפגום, והמחלה תתפתח.
פתוגנזה
בכל סוג של מחלת שארקו-מארי-טות', הפתוגנזה שלה נגרמת על ידי אנומליה תורשתית של עצבים היקפיים: מוטוריים (תנועה) ותחושתיים (רגישים).
אם סוג ה-CMT הוא דה-מיאלינציה, אזי הרס או פגם של מעטפת המיאלין המגנה על האקסונים של העצבים ההיקפיים מובילים להאטה בהעברת דחפים עצביים במערכת העצבים ההיקפית - בין המוח, השרירים ואיברי החישה.
בסוג האקסונלי של המחלה, האקסונים עצמם מושפעים, דבר המשפיע לרעה על עוצמת אותות העצבים, שאינו מספיק לגירוי מלא של שרירים ואיברי חישה.
קראו גם:
כיצד מועברת תסמונת שארקו-מארי-טות'? הגנים הפגומים יכולים לעבור בתורשה באופן אוטוזומלי דומיננטי או אוטוזומלי רצסיבי.
הסוג הנפוץ ביותר, תורשה אוטוזומלית דומיננטית, מתרחש כאשר יש עותק אחד של הגן המוטנטי (נישא על ידי אחד ההורים). וההסתברות להעביר את הגן CMT לכל ילד שנולד מוערכת ב-50%. [ 4 ]
בתורשה אוטוזומלית רצסיבית, נדרשים שני עותקים של הגן הפגום (אחד מכל הורה שאינו מראה סימנים כלשהם של המחלה) כדי שהמחלה תתפתח.
ב-40-50% מהמקרים, מתרחשת דה-מיאלינציה תורשתית אוטוזומלית דומיננטית, כלומר CMT סוג 1; ב-12-26% מהמקרים, CMT אקסונלי, כלומר סוג 2. וב-10-15% מהמקרים, נצפית תורשה מקושרת ל-X. [ 5 ]
תסמינים מחלת שארקו-מארי-טות'
בדרך כלל, הסימנים הראשונים של מחלה זו מתחילים להופיע בילדות ובגיל ההתבגרות ומתפתחים בהדרגה לאורך החיים, אם כי התסמונת עשויה להתבטא בהמשך. שילוב התסמינים משתנה, וקצב התקדמות המחלה, כמו גם חומרתה, בלתי אפשרי לחיזוי.
תסמינים אופייניים לשלב הראשוני כוללים עייפות כללית מוגברת; ירידה בטונוס (חולשה) של שרירי כפות הרגליים, הקרסוליים והשוקיים; חוסר רפלקסים. מצב זה מקשה על תנועת כף הרגל ומוביל לדיסבאזיה (הפרעה בהליכה) בצורת זקפה גבוהה יותר של הרגליים, לעיתים קרובות עם מעידות ונפילות תכופות. סימנים למחלת שארקו-מארי-טות' אצל ילד קטן עשויים לכלול מגושמות בולטת וקשיי הליכה שאינם אופייניים לגיל, הקשורים לצניחת כף רגל דו-צדדית. עיוותים בכף הרגל אופייניים גם הם: קשת גבוהה (כף רגל חלולה) או שטוחה קשות, אצבעות רגליים מעוקלות (בצורת פטיש).
במקרה של הליכה על קצות האצבעות על רקע היפוטוניה שרירית, נוירולוג עשוי לחשוד כי לילד יש CMT מסוג 4, שבו ילדים עשויים להיות חסרי יכולת ללכת עד גיל ההתבגרות.
ככל שהמחלה מתקדמת, ניוון שרירים וחולשה מתפשטים לגפיים העליונות, מה שמקשה על ביצוע מוטוריקה עדינה וביצוע משימות ידיים רגילות. ירידה בתחושות המישוש וביכולת לחוש חום וקור, כמו גם נימול בכפות הרגליים ובידיים, מצביעים על נזק לאקסונים של עצבי החישה.
במחלת שארקו-מארי-טות' מסוגים 3 ו-6, המתבטאת בילדות, נצפית אטקסיה תחושתית (פגיעה בתיאום תנועות ושיווי משקל), עוויתות ורעד שרירים, נזק לעצב הפנים, ניוון עצב הראייה עם ניסטגמוס ואובדן שמיעה.
בשלבים מאוחרים יותר, ייתכנו רעד בלתי נשלט (רעד) והתכווצויות שרירים תכופות; בעיות בתנועה עלולות להוביל להתפתחות כאבים: שרירים, מפרקים, נוירופתיים.
סיבוכים ותוצאות
למחלת שארקו-מארי-טות' יכולים להיות סיבוכים והשלכות כגון:
- נקעים ושברים תכופים יותר;
- חוזים הקשורים לקיצור של שרירים וגידים פריארטיקולריים;
- עקמת (עקמומיות עמוד השדרה);
- בעיות נשימה - כאשר סיבי העצבים המעצבבים את שרירי הסרעפת ניזוקים:
- אובדן היכולת לנוע באופן עצמאי.
אבחון מחלת שארקו-מארי-טות'
האבחון כולל בדיקה קלינית, אנמנזה (כולל היסטוריה משפחתית), בדיקה נוירולוגית ומערכתית.
בדיקות מבוצעות לבדיקת טווח תנועה, רגישות ורפלקסים של גידים. ניתן להעריך הולכה עצבית באמצעות אבחון אינסטרומנטלי - אלקטרומיוגרפיה או אלקטרונוירומיוגרפיה. ייתכן שיידרשו גם אולטרסאונד או MRI. [ 6 ]
בדיקות גנטיות או בדיקות DNA לגילוי המוטציות הגנטיות הנפוצות ביותר הגורמות ל-CMT בדגימת דם מוגבלות מכיוון שבדיקות DNA אינן זמינות כיום לכל סוגי המחלה. למידע נוסף, ראו בדיקות גנטיות.
במקרים מסוימים, מבוצעת ביופסיה של העצב ההיקפי (בדרך כלל העצב הסוראלי).
אבחון דיפרנציאלי
אבחנה מבדלת כוללת נוירופתיות פריפריאליות אחרות, ניוון שרירים על שם דושן, תסמונות מיאלופתיות ומיאסטניות, נוירופתיה סוכרתית, מיאלופתיות בטרשת נפוצה ואמיוטרופית צידית, תסמונת גילאן-בארה, טראומה לעצב הפרונאלי ואטרופיה שלו (כולל כאשר הוא צבט בין הדיסקים המותניים של עמוד השדרה), נזק למוח הקטן או לתלמוס, וכן תופעות לוואי של כימותרפיה (במהלך טיפול בציטוסטטיקה כגון וינקריסטין או פקליטקסל). [ 7 ]
למי לפנות?
יַחַס מחלת שארקו-מארי-טות'
כיום, הטיפול במחלה תורשתית זו כולל טיפול גופני (שמטרתו חיזוק ומתיחה של שרירים); ריפוי בעיסוק (המסייע לחולים עם חולשת שרירים בידיים); ושימוש במכשירים אורתופדיים כדי להקל על ההליכה. במידת הצורך, נרשמים משככי כאבים או נוגדי פרכוסים. [ 8 ]
במקרים של כף רגל שטוחה חמורה, ניתן לבצע אוסטאוטומיה, ובמקרה של עיוות עקב, מומלץ לבצע תיקון כירורגי - ארתרודזיס. [ 9 ]
מחקרים נערכים הן על המרכיב הגנטי של המחלה והן על שיטות הטיפול בה. השימוש בתאי גזע, הורמונים מסוימים, לציטין או חומצה אסקורבית טרם הניב תוצאות חיוביות.
אבל בזכות מחקרים עדכניים, משהו חדש אכן עשוי להופיע בטיפול במחלת שארקו-מארי-טות' בעתיד הקרוב. לפיכך, מאז 2014, החברה הצרפתית Pharnext מפתחת, ומאמצע 2019, מתקיימים ניסויים קליניים עבור התרופה PXT3003 לטיפול ב-CMT סוג 1 במבוגרים, אשר מדכאת ביטוי מוגבר של הגן PMP22, משפרת את המיאלינציה של עצבים היקפיים ומקלה על תסמינים עצביים-שריריים.
מומחים מחברת הרפואה Sarepta Therapeutics (ארה"ב) עובדים על יצירת ריפוי גנטי למחלת Charcot-Marie-Tooth מסוג 1. טיפול זה ישתמש בנגיף אדנו-קשור (AAV) בלתי מזיק מסוג Dependovirus עם גנום DNA חד-גדילי ליניארי, אשר יעביר את הגן NTF3 לגוף, המקודד את חלבון הנוירוטרופין-3 (NT-3) הדרוש לתפקוד תאי העצב של שוואן.
חברת Helixmith תתחיל בניסויים קליניים של ריפוי הגנים Engensis (VM202) שפותח בדרום קוריאה עד סוף 2020 לטיפול בתסמיני שרירים ב-CMT מסוג 1. [ 10 ]
מְנִיעָה
מניעת CMT יכולה להיות ייעוץ גנטי להורים עתידיים, במיוחד אם למישהו מבני הזוג יש היסטוריה משפחתית של המחלה. עם זאת, זוהו מקרים של מוטציות גנטיות נקודתיות דה נובו, כלומר, בהיעדר המחלה בהיסטוריה המשפחתית.
במהלך ההריון, ניתן לבדוק את ההסתברות למחלת Charcot-Marie-Tooth אצל הילד העתידי באמצעות ביופסיה של הסיסי הכוריונית (משבוע 10 עד 13 להריון), וכן ניתוח של מי השפיר (בשבוע 15-18).
תַחֲזִית
באופן כללי, הפרוגנוזה עבור סוגים שונים של מחלת שארקו-מארי-טות' תלויה בחומרתה הקלינית, אך בכל המקרים המחלה מתקדמת באיטיות. לחולים רבים יש מוגבלויות, אם כי הדבר אינו מקטין את תוחלת החיים.